Octad Portal Frequently Asked Questions
Main Idea
What is sRGES?
RGES (Reversal Gene Expression Score), a measure modified from the connectivity score developed in other studies. To compute the RGES score, we first rank genes based on their expression values in each drug signature. An enrichment score/s for each set of up- and down-regulated disease genes are computed separately using a Kolmogorov–Smirnov-like statistic, followed by the merge of scores from both sides (up/down). The score is based on the amount to which the genes (up or down-regulated) at either the top or bottom of a drug-gene list ranked by expression change after drug treatment. One compound may have multiple available expression profiles due to having been tested in various cell lines, drug concentrations, treatment durations, or even different replicates, resulting in multiple RGES for one drug-disease prediction. Therefore, we developed a summarization method to mitigate bias and to compute a score representative of the overall reversal potency of a compound to particular cancer. We termed this score summarized RGES (sRGES).
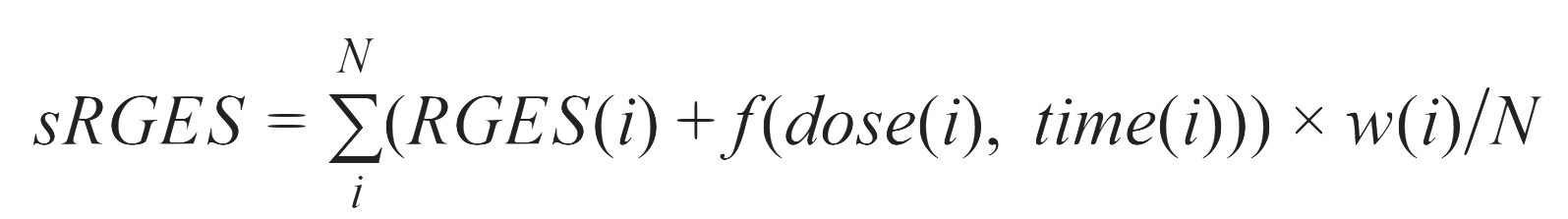
where N is the number of drug profiles. f(dose(i), time(i)) was estimated by a computational model. Correlation between cell(i) and tumour samples was estimated as the average of correlations between the cell line and individual tumours.
For more information, please refer to the original paper: Chen, B., Ma, L., Paik, H., Sirota, M., Wei, W., Chua, M. S., ... & Butte, A. J. (2017). Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nature communications, 8(1), 1-12.
What does the acronym OCTAD stands for?
Open Cancer TherApeutic Discovery
What does OCTAD do?
The goal of OCTAD is to expedite cancer research for therapies. OCTAD allows users to select their cancer or subtype of interest and see which genes and pathways are affected due to the disease. With the LINCS database, we can explore further by predicting potential drugs or targets that may be beneficial against that specific cancer (PMID:28699633). Further evaluation of the drug hits may require bench research for efficacy and/or toxicity testing. Because it is a predictive model, use at your own risk.
How do I use OCTAD?
OCTAD is very easy to use. After picking your cancer and filtering down for desired features, you can either manually choose your control samples (coming soon) or use the given default choice that is most correlated to your cancer tissue type. Choose a method of differential expression in the Signature tab and filter down results with the adjusted P value and the number of fold change seen between the case and control samples. Under the Drugs tab, you can choose which database you want to evaluate the potential to reverse the disease signature expression. You can either compute disease signature earlier or submit the job directly. OCTAD will compute disease signature if it detects there is no signature computed. It takes a few minutes to compute disease signature and about ten minutes to run drug prediction. You can run drug prediction anonymously or through a registered account. A unique URL is created for each job. The files from the anonymous submission will be deleted after three months. A full summary report will be available to review and all data files (description of datafiles is available here) can be downloaded after the job is complete. You can manage your jobs and check job status by going to the jobs page.
Databases
What are the database sources used in OCTAD?
OCTAD uses patient samples from GTEx, TCGA, St. Jude Hospital, MET 500, and TARGET. Please see our Dataset tab for more information about each data source. Our database currently comprises a total of 19128 normal and tumor samples.
Will the databases be continually updated on OCTAD? Will more samples be added into the database later on?
Yes. In future version, we plan to include more patient samples. Please contact us if you have datasets you would like to add into OCTAD.
How frequent do you update the database?
Depends. We have covered the majority of cancers, we will update new large datasets when they are available.
Are there patterns to the naming of sample IDs?
Yes, there are a few patterns. Note that these are the original sample IDs from our sources which have their own naming system. For example, from the GTEx source, the samples are name as GTEX-XXXXX-YYYY-SM-ZZZZZ. XXXXX is the patient ID, YYYY is the code for the tissue, and ZZZZZ is the sample aliquot used for sequencing. Please refer to the dataset tab for more info and links to each data source.
Is there a way to differentiate live and postmortem samples?
No, OCTAD does not list which biopsies are postmortem samples. However, the original data sources have that information. GTEx samples are postmortem samples, except some whole blood samples which were collected prior to death.
Sample Selection Tabs When is the sample size too low?
There is no absolute threshold. If the sample size is low, you may not be able to get signature genes that warrant a good prediction.
After filtering the database for samples, my sample size n is less than 5, what should I do?
If you could not get enough signature genes (> 100), you may need to change your hypothesis.
Where are the genomic features (mutation, copy number variation) from?
We compiled the list from cbioportal. Currently we only cover TCGA samples and include others soon.
How do I interpret the cancer plot? What are the X and Y axes?
This feature is deprecated. This t-SNE plot is to show similarity between samples. The closer the points are, the more similar they are in mRNA expression.
If the distance between points on the t-SNE plot shows similarity, why do I have a few points relatively far from the main island, even though they are same type of tissue?
This feature is deprecated. Those points are either outliers or the samples are mislabeled in the data source. You can remove that sample from your sample set if it is deemed necessary.
I have my own control samples. How do I use my own dataset?
Currently, we DO NOT support users to upload their own data. This function will become available in the next version. Or use the octad package for customized samples.
Can I compare two sample types that are not the cancer-normal pairing? Ie. How would I explore the different expression between two cancer types such as colon cancer and rectum cancer? How do I compare adjacent tissue with normal tissue?
The portal does not support at this moment. Alternatively, you make a prediction for each cancer or subtype and then merge the results. The octad R package supports such advanced queries.
How do I manually choose my samples?
In the sample list, you can manually select samples by checking the ones you want and unchecking the ones you want to exclude in your set.
How do I select more than one type of tissue for control samples?
This feature is not supported in the web portal. Please try OCTAD desktop version which provides more flexibility to choose samples.
Signature Tab
What is the difference between edgeR and Limma under DE (differential expression) method?
They used different methods to compute DE. Please refer to their publications for further explanation.
Drugs Tab
What are the differences between the LINCS databases L1000 and L1000 All?
L1000 uses only 978 landmark genes in the prediction (other 22 genes are used as control). L1000 All include the imputed expression of the remaining transcriptome. By default, L1000 is used.
What number should I input for “max genes on One side”? And what does it mean?
Sometimes, we have to limit the number of disease signature to compute reversal correlation. Empirical analysis suggested 50 is good for L1000.
Results
How long does it take for OCTAD to complete the results of a job?
About 10-20mins.
Miscellaneous
I am interested in being part of your open project. Does your group accept volunteers?
Yes. If you are interested in volunteering please conatact us at octad.contact@gmail.com.
How do I support the efforts of OCTAD?
Please contact us via octad.contact@gmail.com.
How can I add RNAseq data to your tissue sample database?
Yes, but we have to do it manually.
Can I have access to the raw RNA sequence data?
As our pipeline uses sources from other databases, we do not release the raw sequences.
What browsers does OCTAD support?
Chrome, Safari, Microsoft Edge, and Mozilla Firefox. Octad is currently unavailable on mobile devices.
Can I use the results and images in a academic paper?
Yes. The job id is the permanent id. We encourage you to log in before job submission.
What is the desktop version of OCTAD and where can it be accessed?
The backend of the web and desktop versions is done with the R statistical programming language. The desktop version provides more flexibility than the web version. It is a open source project that still is under development. If you have programming knowledge and would like to contribute then check out the GitHub repository at https://github.com/Bin-Chen-Lab/octad_desktop.
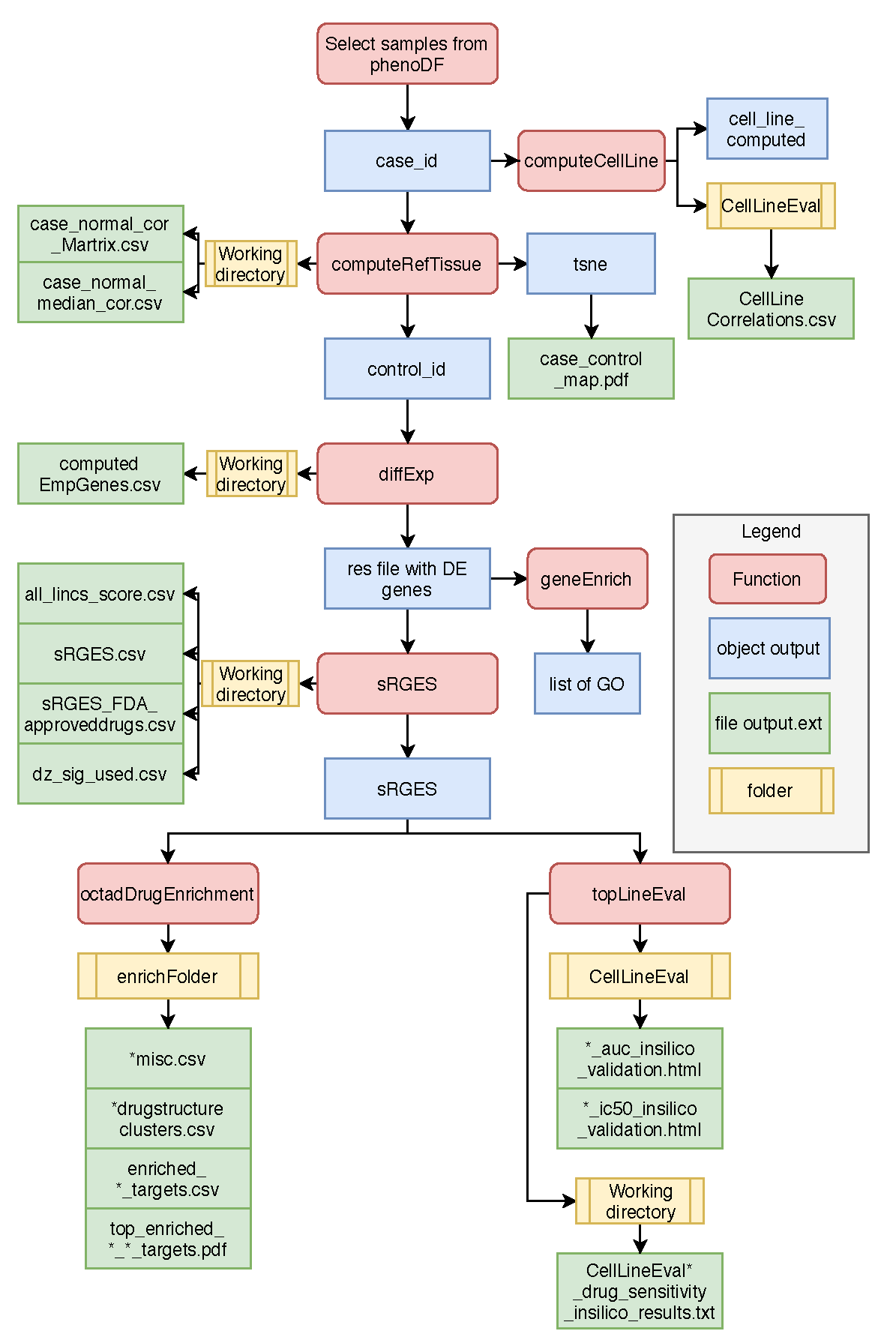
Overall pipeline
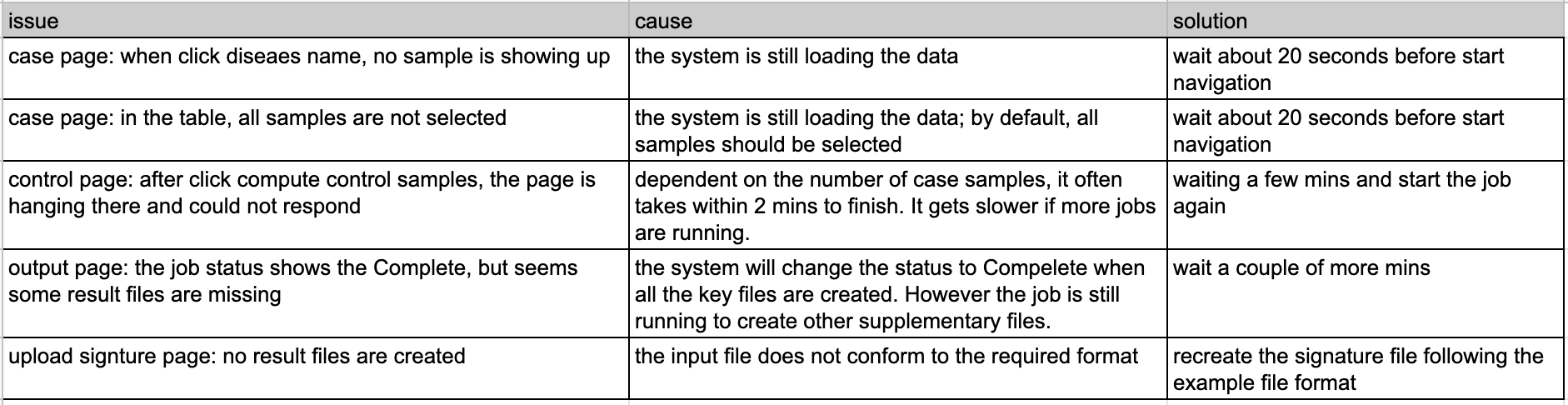
Troubleshooting in web portal
Troubleshooting in R package
| Function | Message | Possible cause | Solution |
|---|---|---|---|
| computeRefTissue | cannot open file 'as//.../case_normal_corMatrix.csv': No such file or directory | outputFolder | Name of the output folders is incorrect. Replace it with valid name. |
| Error in apply(expSet_normal, 1, stats::IQR) : dim(X) must have a positive length | source | Whether source option was modified, no additional matrix was used. Make sure the expSet option is set to custom expression matrix object name | |
| Error in expSet[, normal_id] : incorrect number of dimensions | expSet | Custom matrix input has not enough samples for computation or missing case samples in expression matrix. Make sure after filter out case samples there are still samples. | |
| diffExp | Expression data not sourced, please, modify expSet option | expSet | User decided to use custom file input but not sourced an object. Make sure an expSet option is defined with object name containing expression data |
| Please, source case ids and control ids vector | case_id or control_id | Either case or source vector is not sourced | |
| empty output | annotate | If TRUE, annotation by ENSEMBL gene would be performed. If TRUE, make sure row.names of the custom input contain ensembl gene ids. | |
| Error in h5checktypeOrOpenLoc(file, readonly = TRUE, native = native) : Error in h5checktypeOrOpenLoc(). Cannot open file. File 'octad.counts.and.tpm.h5' does not exist. | source | if source='octad.whole', function estimates octad.counts.and.tpm.h5 to be stored in working directory or full path sourced through file option | |
| Function | Message | Possible cause | Solution |
| runsRGES | Disease signature input not found | dz_signature | Source disease signature with columns Symbol and log2FoldChange |
| Either Symbol or log2FoldChange collumn in Disease signature is missing | dz_signature | Source disease signature with columns Symbol and log2FoldChange | |
| Warning message: In dir.create(outputFolder) : cannot create dir '', reason 'Invalid argument' | OutputFolder | Wrong name of output folder | |
| computeCellLine | Case ids vector input not found | case_id | Case vector is not sourced |
| octadDrugEnrichment | Error in file(file, ifelse(append, "a", "w")) : cannot open the connection | enrichFolder | Wrong name of output folder |
| sRGES input not found | sRGES | sRGES is not sourced | |
| Error in .local(expr, gset.idx.list, ...) : No identifiers in the gene sets could be matched to the identifiers in the expression data. | sRGES | Make sure column pert_iname column not empty | |
| Either sRGES or pert_iname collumn in Disease signature is missing | sRGES | Make sure sRGES tibble input contains both pert_iname and sRGES columns | |
| topLineEval | Error in [.data.frame(x, r, vars, drop = drop) : undefined columns selected |
topline | Make sure your cell lines vector is valid. You can compare output with computeCellLine output |